Chu is a computational biologist dedicated to uncovering the roles of genomic repeats—including transposable elements (TEs), short and long tandem repeats, and large satellite repeats—in human health and disease.
He earned his Ph.D. in Computer Science and Engineering from the University of Connecticut and completed postdoctoral training in the Department of Biomedical Informatics at Harvard Medical School. Before joining The Wistar Institute in 2025, Chu was a principal scientist at the biotechnology company ROME Therapeutics.
The Chu Laboratory

The Chu Laboratory
Genomic repeats make up over half of the human genome and play critical roles in evolution and development. Many diseases have been directly or indirectly linked to these elements. Yet, despite their importance, genomic repeats were historically dismissed as “junk DNA” and remain underexplored. The Chu Lab integrates systems biology with cutting-edge genomics to investigate how genomic repeats influence human health and disease, with a focus on uncovering underlying mechanisms and identifying biomarkers and therapeutic targets. We develop computational methods to quantify genomic repeats from large-scale datasets, enabling detailed analyses of their activity across developmental stages and disease contexts.
Working closely with biologists and clinicians, we aim to address three key questions: (1) What mechanisms has the human body evolved to suppress the activity of genomic repeats? (2) How do genomic repeats evade these defenses—for example, by promoting dsRNA- and dsDNA-derived inflammation—and what roles do they play in disease states? (3) Can these elements be harnessed as biomarkers or therapeutic targets based on these insights?
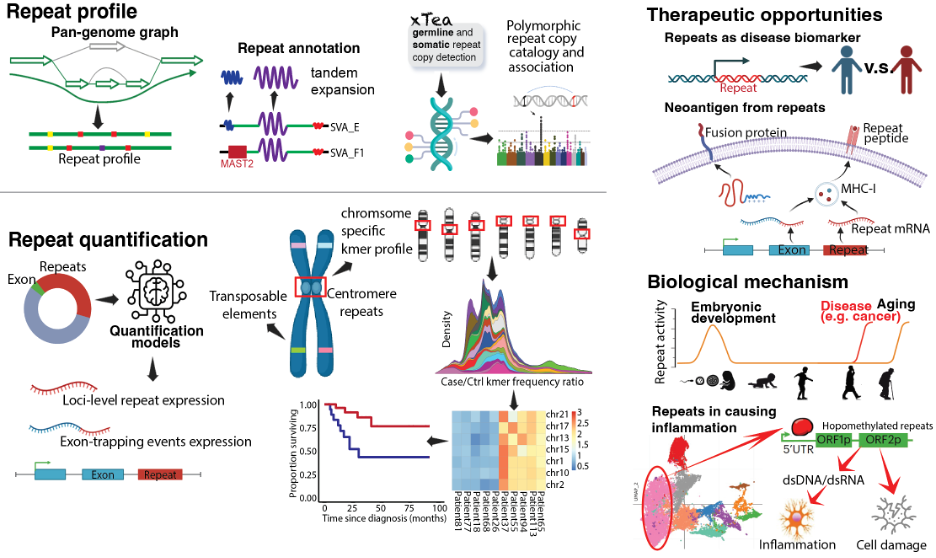
Research
Genomic repeats and their contribution to human health and disease
Genomic repeats are dynamic elements that influence development, evolution, and disease. Accurately detecting these sequences across diverse sequencing technologies has long been a challenge. We developed xTea, a versatile computational pipeline for identifying germline, somatic, and mosaic transposable element (TE) insertions from short-read, linked-read, and long-read whole-genome sequencing data. Using xTea, we identified hundreds of de novo TE insertions in large pediatric cohorts, linking them to conditions such as birth defects and autism spectrum disorder. Building on this foundation, we will leverage long-read sequencing and multi-omics technologies to create robust methods for quantifying repeat activity at the RNA, epigenetic, and proteomic levels—ultimately aiming to map the full spectrum of repeat activity and define their contributions to human health and disease progression.
Algorithm design and method development in computational biology
Our research focuses on creating computational tools that expand the boundaries of genomic discovery. In addition to xTea, we have developed a suite of widely adopted methods—including REPdenovo, GINDEL, SpliceJumper, GAPPadder, and TrapHunter—applied across diverse species and research contexts. For example, REPdenovo has been used to construct repeat libraries for non-model organisms such as the strawberry poison frog and the giant squid, while GINDEL and SpliceJumper were among the earliest applications of machine learning to variant genotyping and splicing analysis. Building on this foundation, we will continue to design scalable algorithms for complex genomic analyses, integrate multi-omics data, and embed advanced machine learning techniques into our pipelines to address questions that lie beyond the reach of conventional methods.
Disease biomarker and early drug discovery
Transposable elements (TEs) are normally silenced through epigenetic mechanisms, but in many cancers—particularly epithelial tumors—they become aberrantly reactivated, producing tumor-specific transcripts and proteins. Our collaborative work has demonstrated that these reactivated TEs can serve both as therapeutic targets and as sources of highly specific tumor antigens. Building on these findings, we will systematically map the landscape of TE-derived biomarkers and drug targets, aiming to develop precision diagnostics and immunotherapies that harness the unique biology of these repetitive elements.
Applied machine learning in understanding biological mechanisms
Machine learning has become indispensable for interpreting the complexity of genomic data. We have integrated data-driven models into diverse projects—from classifying splice junctions in RNA-seq data to identifying and genotyping transposable element (TE) insertions. These approaches have consistently improved accuracy, scalability, and discovery power. Building on this foundation, we are developing and refining machine learning frameworks to elucidate how genomic repeats interact with regulatory networks, modulate immune responses, and shape disease phenotypes. By combining algorithmic innovation with biological insight, we aim to reveal predictive patterns that can drive experimental validation and inform the development of new therapeutic strategies.
